의료기기(용품) 인허가를 위한 대략적인 시험 비용




의료기기를 개발하거나 수입·유통하기 위해서는 다양한 시험을 거쳐 안전성과 성능을 입증해야 합니다. 특히 국내에서 허가를 받기 위해 요구되는 시험 항목들은 매우 다양하며, 이에 따른 비용도 상당합니다.
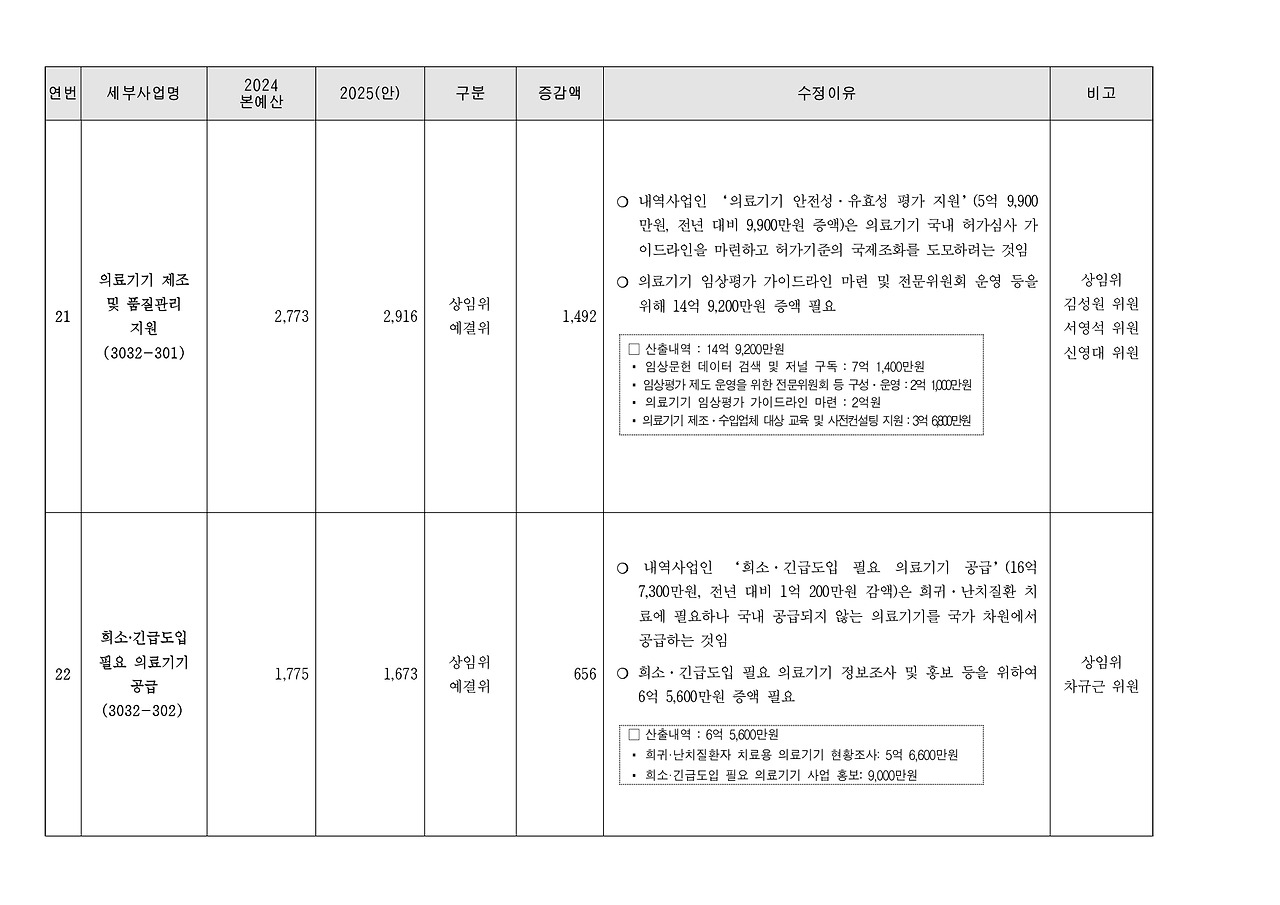
우선 생물학적 시험 패키지는 의료기기의 인체 접촉 여부에 따라 필수적으로 요구되는 시험입니다. 혈액적합성 및 체내이식을 제외한 항목 기준으로 약 1,500만 원에서 2,500만 원 정도의 비용이 발생하며, 세포독성, 피부감작성, 세균성 발열물질 시험 등 다수의 항목이 포함됩니다. 특히 ISO 10993 시리즈를 기반으로 한 이 시험군은 기기의 사용 부위와 기간에 따라 그 범위와 난이도가 달라지므로, 초기 계획이 중요합니다.
멸균 유효성 시험은 멸균처리된 의료기기에 필수적으로 적용됩니다. EO(에틸렌옥사이드) 멸균, 방사선 멸균 등 멸균 방식에 따라 시험 방법이 다르며, 시험 비용은 약 1,000만 원에서 2,000만 원 선으로 형성되어 있습니다. 이는 멸균 공정의 반복성, 멸균 보증 수준(SAL) 입증 등에 따른 시험이 포함되기 때문입니다.
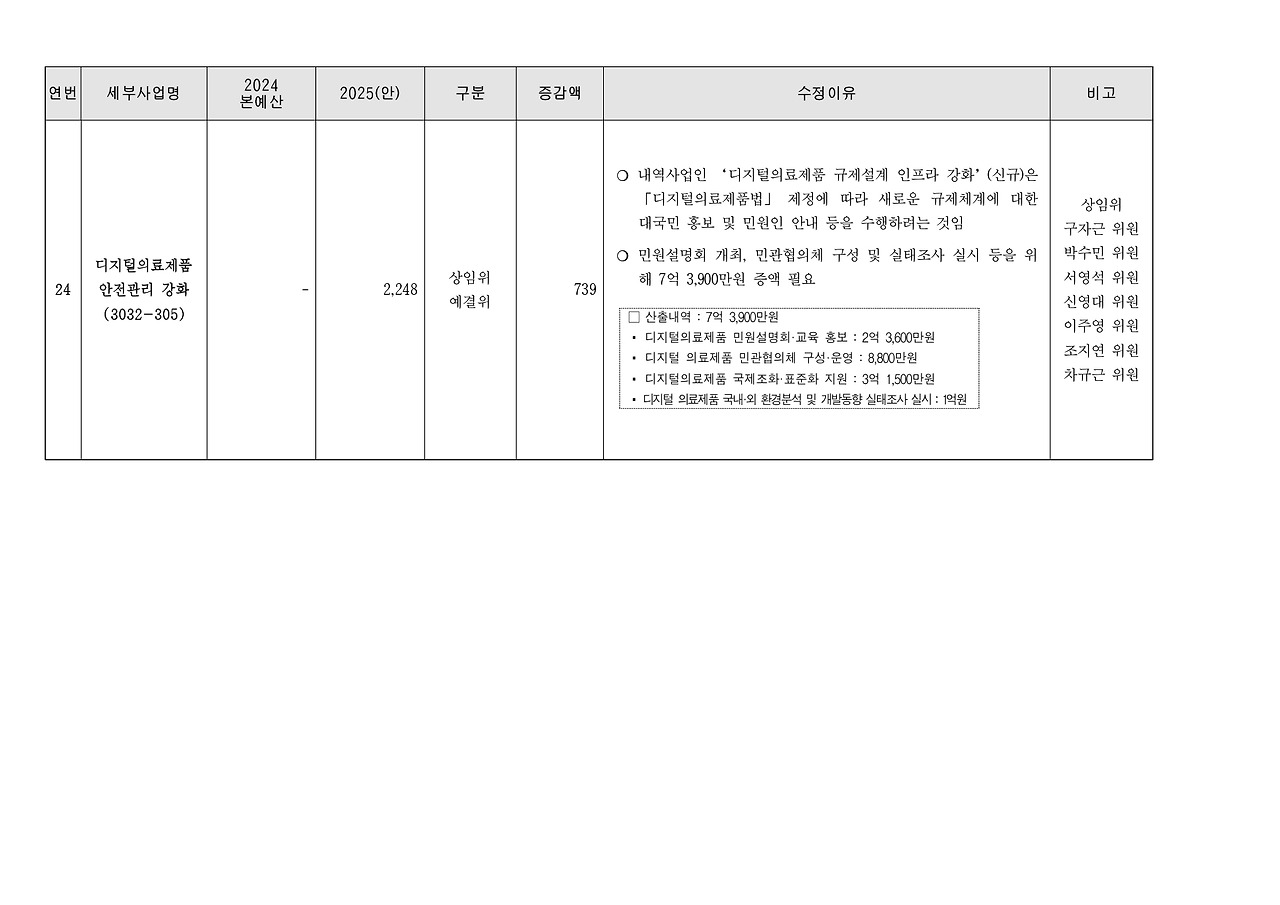
멸균 공정에서 흔히 사용되는 EO 잔류 시험은 EO 멸균 후 인체에 유해할 수 있는 EO 가스의 잔류량을 측정하는 시험으로, 약 150만 원에서 300만 원 정도의 비용이 소요됩니다. 이 시험은 ISO 10993-7 기준에 따라 EO, ECH, EG 등 잔류물질의 정량분석을 수행합니다.
포장 무결성 및 가속노화 시험은 의료기기의 유효기간 설정 및 운송·보관 안정성 확보를 위한 시험입니다. 포장재가 물리적 충격이나 환경 변화에도 무결하게 기능을 유지하는지를 입증해야 하며, 이로 인한 비용은 약 300만 원에서 500만 원 수준입니다. 가속노화 시험을 통해 유효기간 동안 성능이 유지되는지 예측하는 것도 이 항목의 주요 목적입니다.
마지막으로, 물리적/성능 시험은 제품의 기계적 안정성, 작동 특성, 전기적 안전성 등을 평가하는 항목으로, 약 100만 원에서 300만 원의 비용이 발생합니다. 해당 시험은 제품의 유형에 따라 그 범위가 달라지며, IEC 또는 KS 기준을 기반으로 시험이 이루어집니다.
'의료기기 RA > 의료기기 인허가' 카테고리의 다른 글
| 미용기기 및 경계성 의료기기의 안전한 규제와 유통 관리의 중요성 (0) | 2025.07.20 |
|---|---|
| 만성질환 관리 및 시·청각장애인 사용 의료기기 예시 (0) | 2025.05.13 |
| 의료기기 규제 강화를 위한 대한민국 정부 2025년 식약처 예산 방향 (0) | 2025.04.30 |
| 2등급 의료기기 기술문서심사 수수료 인상에 대한 고찰 (0) | 2025.04.21 |
| FDA 허가, 의료기기 상업화의 시작일 뿐 (0) | 2025.04.20 |